
By Peyton Howell
The pharma industry has begun a new chapter of the biosimilars story. We now have much-anticipated guidance from the FDA on what the approval pathway will look like, what evidence can be used from a branded innovator, and a sense of how cumbersome the approval process might be for manufacturers of biosimilars. Based on the guidance, the process of bringing a biosimilar to market will likely require many of the strategies used to bring a branded product to market, which will impact the commercialization of the products and likely reduce potential cost savings.
The explosive growth in the costs associated with pharmaceuticals, particularly highly complex specialty products, continues to be a significant barrier to curbing overall healthcare spending in the United States. As such, biosimilars are expected to decrease or at least control rising pharmaceutical expenditures by offering considerable savings for high-cost biologic products. According to the Congressional Budget Office, biosimilars are expected to save $13 billion over the next 10 years. Initially, from the payer perspective, biosimilars were expected to generate 30% to 40% savings; however, as the complexity of bringing biosimilars to market continues to be discussed, more recent data suggest that most payers anticipate a biosimilar will come in between 10% to 20% less than the cost of the branded manufacturer. In Europe, we know that biosimilars have often been just a 10% discount from the brand. Figure 1 illustrates trending payer opinions based on Xcenda's proprietary research with advisers who participate in a managed care network. There has been a significant shift from payer perception on savings between 2010 and 2011.
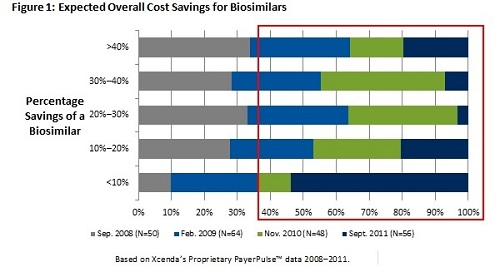
The premise of biosimilar cost savings is that the FDA approval pathway is significantly less arduous than that of a branded biologic. In November 2010, the FDA held a public meeting on biosimilars, which included representatives from both branded and generic manufacturers. Not surprisingly, branded manufacturers cited the need for independent evidence and strong data to support approval by the FDA. On the contrary, generic and potential biosimilar manufacturers identified that the purpose of such a pathway is to ensure products can be approved based on innovator data and in a reduced time frame to improve access to lower-cost products.
On Feb. 9, the FDA released the first substantive information since the passage of the ACA in the form of three guidance documents that provide some insight into the approval pathway for biosimilars. These regulatory guidances suggested some wins for biosimilar manufacturers, as the FDA clarified that some ex-U.S. and branded innovator data can be used to support a biosimilar application for approval. Additionally, the FDA suggested that biosimilar manufacturers will be allowed to extrapolate innovator data to support the approval of all indications the branded product is approved for. While these may reduce the overall resources required to bring a biosimilar to market, there were other aspects of the guidance that were not as favorable to these stakeholders. For example, the FDA discussed some methodological approaches to examine interchangeability; however, the guidance suggests that the FDA does not believe the current technology is evolved enough to truly establish interchangeability.
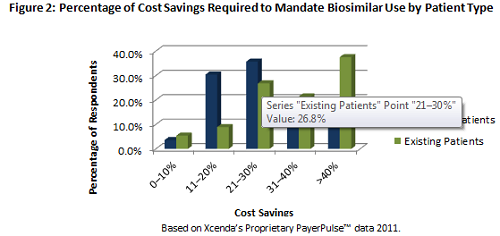
While these documents provide some clarity from the FDA regarding evidence aspects of the approval pathway, the lingering issue with interchangeability may prove to be a barrier for biosimilar manufacturers from the utilization and payer management perspectives. Based on Xcenda research, in order to require an existing patient to switch to a biosimilar:
-- Payers will want to see greater than a 40% cost savings (see Figure 2) for an existing patient to require a switch
-- Interchangeability will be a key factor in requiring a switch
-- The savings is lower for a new start patient (21% to 30%), indicating that payers may require biosimilar use for new patients sooner in the treatment continuum (see Figure 2)
Physician groups at the 2010 FDA public meeting indicated that interchangeability will be important to ensure uptake and use of biosimilars, particularly the first few biosimilars to market. Because the FDA has yet to fully detail how and if it will establish interchangeability, presumably the bar will be higher than approval only. This, unfortunately, has the potential to drive the development costs up, and thus impact overall healthcare cost savings.
A further consideration from a manufacturer perspective is the type of marketing and patient support investment biosimilars will require to ensure uptake and use. The FDA guidance did not shed any light on other outstanding issues such as the naming, labeling/tracking, and post-marketing monitoring requirements for a biosimilar. As a result of the ambiguity, biosimilar manufacturers looking to bring a product to market within the next 12 to 24 months are likely going to need a pre- and post-launch strategy that incorporates most if not all of the same components as their branded competitors. Billing and coding guidance, value proposition information, and other payer and provider support tools will be necessary to ensure provider uptake and payer coverage. Additionally, while biosimilars are expected to be somewhat lower in cost than their branded equivalent, the patient cost share of these specialty products will likely remain relatively high. Therefore, traditional patient support services (e.g., copay assistance, insurance research assistance) will be essential to ensure use.
While biosimilars provide a promise of healthcare cost savings, the initial impact is still unclear given the need for further FDA guidance (additional guidance is forthcoming from the FDA), challenges around establishing interchangeability, and the broader investment required by manufacturers of biosimilars. However, given the interest across multiple stakeholders for patients to have access to biologic products at a lower cost, the buzz around biosimilars is likely to continue to grow.
Peyton Howell is president of AmerisourceBergen Consulting Services and senior vice president of business development for AmerisourceBergen Corp.